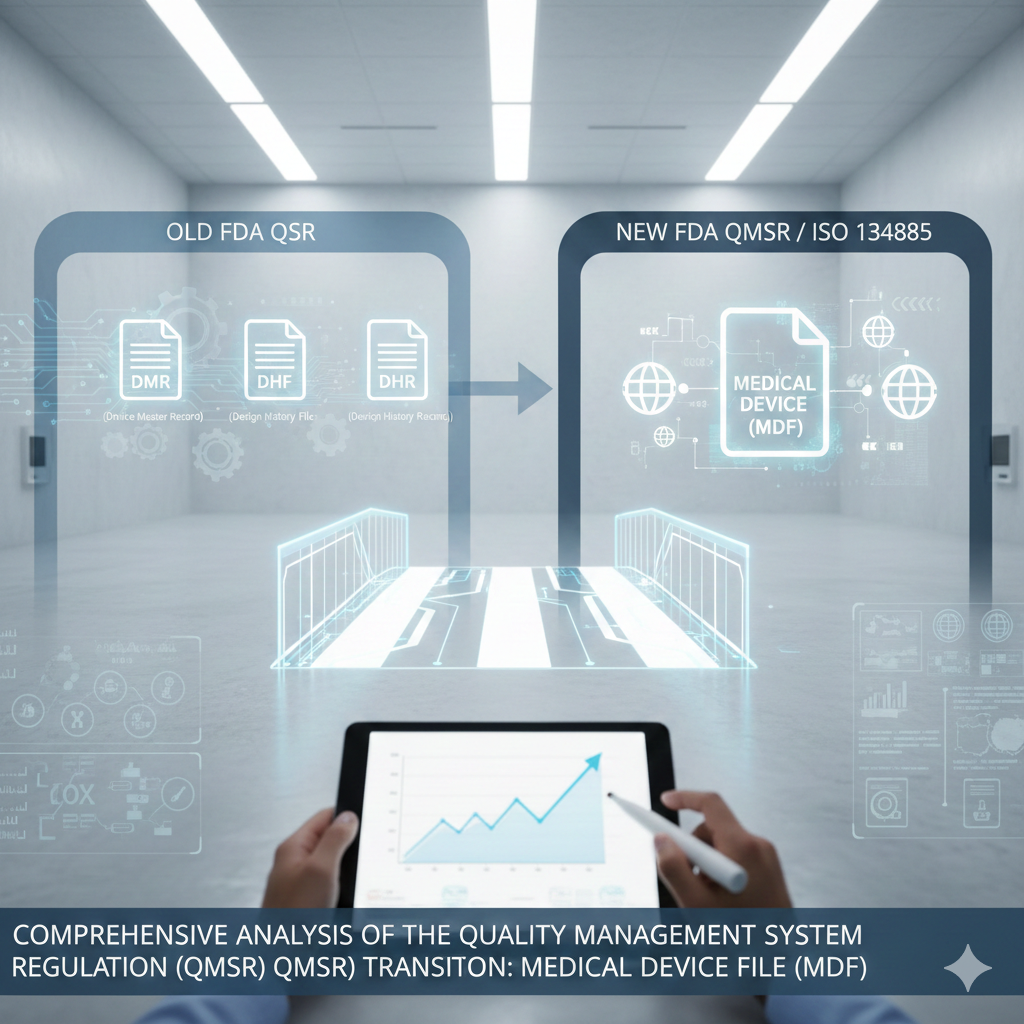
The landscape of medical device regulation in the United States is currently undergoing its most significant transformation since the mid-1990s. On February 2, 2024, the Food and Drug Administration (FDA) issued the “Medical Devices; Quality System Regulation Amendments” final rule, hereafter referred to as the Quality Management System Regulation (QMSR).1 This rule formally incorporates by reference the international consensus standard ISO 13485:2016, “Medical devices—Quality management systems—Requirements for regulatory purposes,” into 21 CFR Part 820.3 This shift is not merely a linguistic update but a fundamental alignment of the American regulatory framework with the global standards utilized by nearly every other major regulatory authority, including those in the European Union, Canada, Japan, and Australia.3 The transition marks the official retirement of legacy documentation constructs—most notably the Design History File (DHF), the Device Master Record (DMR), and the Device History Record (DHR)—in favor of the Medical Device File (MDF) and a risk-based lifecycle approach.6
The Historical Trajectory of Medical Device Quality Oversight
The regulatory journey toward the QMSR began decades ago, rooted in the foundational requirements of the Federal Food, Drug, and Cosmetic Act. Under section 520(f) of the Act, the FDA was granted the authority to establish current good manufacturing practice (CGMP) requirements for medical devices.3 The initial iteration of these requirements, published in 1978 and effective in late 1978, focused primarily on manufacturing controls and final product testing.3 However, as the industry evolved and technological complexity increased, the limitations of a purely manufacturing-centric model became evident. High rates of device recalls were frequently linked to design flaws that occurred long before a product reached the assembly line.8
This realization led to the Safe Medical Devices Act of 1990, which empowered the FDA to mandate preproduction design controls.8 The resulting 1996 revision, known as the Quality System Regulation (QSR), introduced the concept of design controls and established the record-keeping triad of DHF, DMR, and DHR.3 Even during the 1990s, the agency recognized the benefits of international harmonization, participating in the Global Harmonization Task Force (GHTF) and ensuring that the 1996 QSR was consistent, to the extent possible, with ISO 9001:1994 and early versions of ISO 13485.3 For thirty years, this system served as the gold standard for U.S. compliance, but it created a dual-compliance burden for international manufacturers who were forced to maintain disparate quality management systems (QMS) to satisfy both the FDA’s QSR and the ISO 13485 requirements preferred by international notified bodies.5
The evolution from QSR to QMSR represents the fulfillment of a decades-long objective to modernize and harmonize these requirements. The FDA has determined that ISO 13485:2016 is substantially similar to the 1996 QSR in its ability to ensure device safety and effectiveness.4 By adopting the international standard, the FDA aims to reduce redundant regulatory effort, facilitate global market access for domestic manufacturers, and leverage the risk-based process approach that has become the hallmark of modern quality management.5
Architectural Components of the QMSR Framework
The QMSR is structured to integrate the international requirements of ISO 13485:2016 directly into the Code of Federal Regulations, while maintaining several U.S.-specific requirements necessitated by the Federal Food, Drug, and Cosmetic Act.2 The regulation is essentially a hybrid model: it incorporates ISO 13485 by reference but establishes “additional requirements and provisions” that clarify FDA expectations and ensure consistency with other parts of the CFR.1 This structural approach means that while a manufacturer must comply with the clauses of ISO 13485, they must also adhere to specific FDA mandates regarding labeling, packaging, and record availability that the agency believes the international standard does not address with sufficient granularity.2
| Regulatory Element | Source Framework | Application |
| Quality Management System Requirements | ISO 13485:2016 | General QMS, Resource Management, Product Realization.1 |
| Terms and Definitions | ISO 9000:2015 (Clause 3) | Fundamental vocabulary for quality systems.2 |
| Control of Records | 21 CFR 820.35 | Specific requirements for signatures, dates, and accessibility.1 |
| Labeling and Packaging | 21 CFR 820.45 | Prescriptive controls for inspection of labeling and packaging.2 |
| Risk-Based Decisions | ISO 13485:2016 | Explicit requirement for risk management throughout the QMS.2 |
The adoption of ISO 9000:2015 Clause 3 for definitions is particularly critical. This ensures that terms such as “correction,” “corrective action,” and “preventive action” are used consistently with international practice, though the FDA has carefully retained several of its own definitions—such as “manufacturer” and “product”—where it believes the ISO definitions are less comprehensive or incompatible with U.S. law.2
The Retirement of Legacy Records: DHF and DMR
Under the 1996 QSR, the Design History File (DHF) and the Device Master Record (DMR) were the twin pillars of technical documentation. The DHF, mandated by §820.30(j), was intended to demonstrate that a device was developed in accordance with the approved design plan and the requirements of the QSR.17 It served as the chronological narrative of the design journey, capturing user needs, design inputs, verification protocols, and validation reports.20 Conversely, the DMR, mandated by §820.181, was the “master recipe” for the device, containing all specifications, procedures, and instructions required for consistent manufacturing.17
The QMSR eliminates these specific terms from the regulation, replacing them with the structure found in ISO 13485 Clause 4.2.3 (Medical Device File) and Clause 7.3.10 (Design and Development File).6 This change is driven by the agency’s belief that maintaining the legacy terms would be redundant and confusing in a harmonized environment.7 Manufacturers are not prohibited from using the terms DHF and DMR internally, but they will no longer find them in the text of 21 CFR 820.7
The Medical Device File as the Central Production Repository
The Medical Device File (MDF) is defined in ISO 13485 Clause 4.2.3 and serves as a comprehensive, unified repository that consolidates the intent of the DMR and components of the DHF.13 The MDF is required for each device type or device family and must contain or reference all documentation necessary to demonstrate conformity to both the standard and applicable regulatory requirements.22In the transition from a DMR to an MDF, the focus shifts from a static “record” to a dynamic “file” that encompasses the device’s entire current state.22 The MDF acts as a “here and now” snapshot of the device’s compliance, including latest specifications, manufacturing work instructions, and quality assurance procedures.22 It effectively bridges the gap between the EU’s concept of “Technical Documentation” and the U.S.’s traditional manufacturing master records.13
| Legacy Term | QMSR/ISO Equivalent | Key Documentation Transferred |
| Device Master Record (DMR) | Medical Device File (MDF) | Bill of Materials (BOM), drawings, assembly instructions, software source files.6 |
| Design History File (DHF) | Design & Development File (DDF) | Design inputs, verification/validation testing results, design reviews.6 |
| Device History Record (DHR) | Production Records | Batch records, quantity released, acceptance results, labeling used.6 |
The Design and Development File and the Lifecycle Narrative
Clause 7.3.10 of ISO 13485:2016 introduces the Design and Development File (DDF), which mirrors the function of the legacy DHF.6 The DDF must contain or reference all records necessary to prove that the design was developed according to the plan and meets the requirements of the standard.18 This includes the integration of risk management activities, which in a modern QMS are often managed through a Risk Management File (RMF) per ISO 14971.6
The DDF emphasizes the “why” behind the design, preserving the history of design iterations, changes, and the rationale for design choices.19 In a harmonized environment, the DDF and the MDF work in tandem: the DDF documents the evolution of the device, while the MDF contains the finalized outputs that define the current manufacturing state.17
Crosswalk Analysis: Mapping Legacy Records to the QMSR
For organizations that have operated solely under the 1996 QSR, the transition requires a granular mapping of existing procedures and records to the new ISO structure. This “crosswalk” is essential for maintaining compliance and preparing for future inspections.6
Design and Development Transformation
The mapping of the DHF to the DDF is relatively straightforward but requires attention to the more explicit risk management requirements of ISO 13485.14 Under the QMSR, the design outputs that were previously transferred from the DHF to the DMR are now considered part of the MDF product specifications.6 The DDF retains the historical records of the design process, including design plans, user needs, and verification and validation (V&V) evidence.6For software-heavy devices or Software as a Medical Device (SaMD), the DDF mapping must include specific software lifecycle artifacts, such as requirements specifications, architecture diagrams, and testing reports.20 The crosswalk for SaMD also necessitates the inclusion of a Software Bill of Materials (SBOM) and environment snapshots in the production records to ensure traceability of the software build.6
Manufacturing and Production Traceability
The transition from the DHR to the production records required by ISO 13485 Clause 7.5 is more comprehensive than many manufacturers anticipate.6 While the legacy DHR focused on a set list of manufacturing outputs (dates, quantities, labels), the ISO 13485 production record requirements are broader.6 They explicitly demand documentation of the equipment used, process parameters like temperature or speed, and the specific personnel involved in critical steps.6 This shift requires manufacturers to move beyond simple “pass/fail” logs toward detailed records of manufacturing execution.6
The QMSR maintains the requirement that these records demonstrate the device was manufactured according to the specifications in the MDF.7 This relationship ensures that the traceability link between design, manufacturing specifications, and the physical product remains unbroken.17
Technical Amendments Across the Code of Federal Regulations
The publication of the QMSR Final Rule necessitated a massive editorial undertaking by the FDA to ensure consistency across the entire body of medical device regulations.1 The “Technical Amendments” rule, issued alongside the QMSR, modified 179 sections of the CFR spread across 18 parts.1 These changes update internal references to §820, replacing citations to the old QSR with the corresponding sections of the QMSR.1
Impacts on Specialized Regulatory Parts
The amendments touch almost every facet of medical device regulation, from labeling to investigational studies. For example, Part 801, which governs labeling, was updated to change the term “design history file” to “design and development files”.1 Part 803, regarding Medical Device Reporting (MDR), was amended to ensure that references to quality system records are consistent with the new QMSR numbering, particularly shifting from §820.180 (Records) and §820.198 (Complaint files) to §820.35 (Control of Records).1
Part 812, covering Investigational Device Exemptions (IDE), also received technical corrections to ensure that clinical studies are conducted in a manner consistent with the new QMSR expectations, particularly around design controls for investigational products.1 The sheer scale of these amendments underscores the FDA’s commitment to a full systemic alignment with the ISO-based model, ensuring there are no lingering inconsistencies that could lead to regulatory ambiguity.1
Classification Regulation Updates
Many device classification regulations (Parts 862 through 892) historically exempted specific devices from most of the quality system requirements, provided they maintained compliant records and complaint files.1 The technical amendments update these exemptions to reference §820.35 (Control of Records), ensuring that even low-risk, exempt devices remain subject to the fundamental record-keeping and transparency requirements of the QMSR.1
The Transformation of the FDA Inspection Model
The most tangible impact of the QMSR for manufacturers will be the end of the Quality System Inspection Technique (QSIT) and the introduction of a new inspection paradigm.5 Since 1999, QSIT has been the primary tool for FDA investigators, utilizing a subsystem approach (Management, Design, CAPA, Production) to evaluate QMS compliance.2 Effective February 2, 2026, the FDA will withdraw QSIT.12
Implementation of Compliance Program 7382.845
In its place, the FDA is developing a new inspection process that will be documented in a revised version of Compliance Program (CP) 7382.845, “Inspection of Medical Device Manufacturers”.12 This new program will align with the QMSR and the ISO 13485:2016 structure.12 The agency has promised to release this revised CP no later than the effective date of February 2, 2026, providing the field staff with the instructions necessary to audit against the international standard.12
The new inspection model is expected to move away from the siloed subsystem approach of QSIT in favor of a risk-based process approach that evaluates how the various elements of the QMS interact.2 Investigators will look for documented objective evidence of risk-based decisions throughout the device lifecycle, from early product realization through post-market surveillance.14
The Removal of Record Exemptions and the “Full Access” Mandate
A landmark change in the QMSR is the elimination of the record exemptions previously found in 21 CFR 820.180(c).5 Under the 1996 QSR, the FDA generally refrained from inspecting internal audit reports, management review reports, and supplier audit reports.12 The intent was to encourage manufacturers to identify and fix problems without fear that their internal findings would be used against them in enforcement actions.11
Under the QMSR, these records are fully within the scope of FDA inspections.5 The FDA justifies this shift by noting that manufacturers are already required to provide these documents to other regulators and third-party auditors (such as those for MDSAP or ISO 13485 certification).5 Consequently, the agency believes that making these records available to FDA investigators does not impose a new burden.12 For manufacturers, this means that management review minutes and internal audit findings must be maintained with a level of professionalism and rigor suitable for direct regulatory scrutiny.12
Global Convergence: Comparing the MDF with EU MDR Technical Documentation
For manufacturers competing in the global market, the shift to the MDF provides a long-awaited bridge to the requirements of the European Union Medical Device Regulation (EU MDR 2017/745).22 While the EU MDR uses the term “Technical Documentation” (detailed in Annex II and III), the underlying data requirements are highly consistent with the ISO 13485 Medical Device File.22
Convergence of Data Requirements
Both the MDF and the EU Technical Documentation require a comprehensive set of evidence, including device description, specifications, labeling, IFU, risk management files, and clinical evaluation reports.28 For a manufacturer, this means that a single, well-structured “master” technical file can satisfy the core requirements for both the U.S. and the EU, provided that region-specific annexes are added to address local mandates like UDI or EUDAMED registration.22
| Feature | ISO 13485 MDF | EU MDR Annex II/III |
| Objective | Ensure manufacturing consistency and QMS compliance.22 | Demonstrate conformity with GSPRs for CE marking.25 |
| Living Document | Updated as design/process changes occur.22 | Updated through the entire lifecycle and PMS.28 |
| External Review | Inspected by FDA during facility audits.22 | Assessed by Notified Body for certification.25 |
| Post-Market Role | Integrated with QMS feedback loops.14 | Explicit requirement for PMS and PMCF plans.28 |
The primary divergence remains the role of clinical evidence. The EU MDR demands a detailed Clinical Evaluation Report (CER) for all devices, whereas the U.S. focuses on clinical data primarily through the pre-market submission process (510(k) or PMA), while the QMS focuses on ensuring that any clinical validation conducted is properly documented and maintained.16
The Role of Notified Bodies vs. the FDA
While the documentation structure is converging, the oversight mechanism remains distinct. In the EU, Notified Bodies perform a detailed assessment of the Technical Documentation before a product is placed on the market.25 In the U.S., the FDA reviews technical data through pre-market submissions, but the MDF is evaluated primarily during post-market inspections to ensure that the “as-manufactured” device continues to meet the “as-cleared” specifications.16
Risk-Based Lifecycle Management: The Core of the QMSR
The move to ISO 13485:2016 introduces an explicit requirement for a risk-based approach to the entire quality management system, not just design controls.2 This is a significant evolution from the 1996 QSR, which only explicitly mentioned risk analysis within the design validation section (§820.30(g)).9
Integration of ISO 14971
Under the QMSR, manufacturers are expected to apply risk management principles to all QMS processes, including purchasing, supplier controls, training, and production.2 This generally involves the application of ISO 14971, the international standard for medical device risk management.14 The agency now expects manufacturers to provide documented objective evidence of risk-based decisions throughout the product realization process.14
This “risk-based process approach” ensures that resources are allocated effectively, focusing on those areas that have the greatest impact on product safety and patient health.2 For instance, a manufacturer’s supplier audit schedule should be driven by the risk of the component or service provided, rather than a arbitrary calendar-based interval.27
Post-Market Surveillance and Feedback Loops
The QMSR places increased importance on Clause 8 of ISO 13485, which covers monitoring, measurement, and improvement.14 Manufacturers must go beyond reactive complaint handling and implement proactive systems for gathering feedback from the field.14 This proactive data should feed directly back into the risk management process and the design process for future iterations of the product.14
| Lifecycle Phase | Risk Management Activity | Documentation Repository |
| Concept/Design | Hazard identification and risk estimation | Risk Management Plan (DDF).6 |
| Development | Risk control measures and V&V of controls | Risk Management File (DDF/MDF).14 |
| Manufacturing | Process FMEA and process validation | Production Records.6 |
| Post-Market | Analysis of complaints, feedback, and MDRs | PMS Reports (linked to MDF).14 |
Economic Analysis of the QMSR Transition
The shift to the QMSR is expected to have a profound economic impact on the medical device sector. The FDA’s regulatory impact analysis suggests that the rule is a “major” rule under the Congressional Review Act, with significant benefits in the form of cost savings.34
Projected Cost Savings for Establishments
The primary benefit of the QMSR is the reduction of compliance effort for firms that previously had to navigate two different quality system regulations.5 The FDA estimates that the final rule will result in net cost savings of over $500 million for small entities alone.34 These savings stem from the elimination of redundant documentation, the reduction in audit preparation time, and the streamlining of global regulatory submissions.10
The annualized cost savings are estimated to be approximately $540 million at a 7% discount rate.34 While some commenters in the rulemaking process argued that these savings would be negligible in the short term due to the initial costs of training and documentation updates, the FDA maintains that the long-term benefits of harmonization significantly maximize net benefits to the public health and the economy.34
Implementation and Training Costs
The transition is not without its costs. Manufacturers must invest in the initial training of personnel on ISO 13485:2016 and the specific nuances of the QMSR.5 There are also one-time costs associated with updating information technology systems and internal documentation (such as quality manuals and SOPs) to reflect the new structure and terminology.33 The FDA itself will incur costs related to training its vast field staff of investigators to ensure they can effectively and consistently audit against the new standard.12
Strategic Roadmap for Implementation and Compliance
With the enforcement date of February 2, 2026, fast approaching, manufacturers must adopt a systematic approach to QMSR implementation. The transition period is intended to allow for the gradual phasing out of QSR-based systems in favor of the QMSR.1
Phase 1: Strategic Planning and Gap Analysis
Organizations should begin by conducting a comprehensive gap analysis between their current QMS and the requirements of ISO 13485:2016 and the QMSR additions.5 This analysis should focus not just on documentation but on the underlying processes, particularly risk management, competency assessment, and post-market feedback loops.5
Phase 2: Documentation Transition and MDF Establishment
The next phase involves the structural update of the QMS. This includes the creation of a Quality Manual (required by Clause 4.2.2 of ISO 13485) and the formal establishment of the Medical Device File and Design and Development File.13 Manufacturers should decide whether they wish to retain the legacy terms DHF/DMR/DHR internally or move entirely to the ISO terminology; regardless of the choice, a clear crosswalk or transition matrix is necessary for inspection readiness.6
Phase 3: Personnel Training and Competency Assessment
Training is a critical component of the transition. However, the QMSR emphasizes competency over simple training attendance.14 Organizations must move beyond “reading and signing” procedures toward objective assessments of understanding.14 This ensures that personnel are not only aware of the new requirements but possess the skills and knowledge to implement them effectively in their daily tasks.14
Phase 4: Pilot Projects and Internal Audits
Before the 2026 deadline, manufacturers should pilot the new processes on selected product lines.6 Conducting internal audits using the new QMSR framework is essential to identify any remaining gaps in compliance or process maturity.33 These internal audits will be critical practice for the upcoming era of transparent management reviews and audit reports.12
The Future of Global MedTech Regulation
The implementation of the QMSR represents a paradigm shift in how the FDA oversees the quality of medical devices. By moving toward a harmonized, risk-based system, the agency is acknowledging that the quality of medical technology is a global challenge that requires a shared technical and regulatory language.4
The Role of MDSAP in the New Regulatory Era
While the FDA will not require ISO 13485 certification and will not issue certificates of its own, the Medical Device Single Audit Program (MDSAP) remains a valuable tool for manufacturers.12 MDSAP audits already cover the requirements of ISO 13485 and several FDA-specific mandates, making the program a “best indicator” for how the agency will create consistency in its new inspection model.14 Manufacturers currently in compliance with MDSAP will likely find the transition to QMSR inspections virtually seamless.14
Sustaining Quality Through Lifecycle Management
The ultimate goal of the QMSR is to ensure that medical devices are safe and effective through their entire lifecycle, not just at the point of market clearance.13 The shift to the Medical Device File—a living, adaptable record—reflects this focus on continuous quality.22 As medical technologies continue to evolve toward digital health, artificial intelligence, and personalized medicine, the flexible and risk-based framework of the QMSR provides a durable foundation for future innovation.13
The QMSR transition is a watershed moment for the medical device industry, promising to reduce regulatory friction and enhance product quality by aligning the United States with the global community.5 Manufacturers who leverage this two-year transition period to deeply integrate risk-based thinking and modernize their documentation practices will be well-positioned to succeed in this new harmonized regulatory environment.5
Detailed Analysis of Technical Amendments by CFR Part
The complexity of the QMSR transition is most evident in the detailed technical amendments made to the Code of Federal Regulations. The FDA’s decision to modify nearly 180 sections across 18 parts ensures that the shift from QSR to QMSR is reflected in every regulatory interaction a manufacturer may have with the agency.1
| CFR Part | Subject Area | Primary Amendment Type |
| Part 4 | Combination Products | Conforming edits to clarify QMS requirements for constituent parts.12 |
| Part 801 | Labeling | Terminological update from DHF to DDF and record control references.1 |
| Part 803 | Medical Device Reporting | Update of record retention and complaint file references to §820.35.1 |
| Part 812 | Investigational Device Exemptions | Alignment of design control references for investigational products.1 |
| Part 820 | Quality Management System | Fundamental rewrite to incorporate ISO 13485 by reference.1 |
| Part 860 | Device Classification | Updates to exemptions and references to quality system controls.1 |
The amendments in Part 801 (Labeling) are particularly noteworthy. The agency has replaced the specific requirement for a Design History File with the more general requirement for “design and development files” as defined in ISO 13485.1 Similarly, in Part 803 (MDR), the agency has unified the disparate record-keeping requirements for complaints and adverse events under the broader “Control of Records” requirements of §820.35.1 This simplification reduces the administrative burden of tracking multiple, overlapping record-retention mandates.1
The Impact on Premarket Submissions: PMA and HDE Guidance
In October 2025, the FDA issued a draft guidance titled “Quality Management System Information in Premarket Submissions,” which provides specific instructions on how QMSR expectations should be reflected in Premarket Approval (PMA) and Humanitarian Device Exemption (HDE) applications.16 This guidance signals a major departure from the legacy, U.S.-centric submission format.16
Mapping Submission Content to ISO 13485 Clauses
Under the new guidance, the FDA recommends that applicants organize their QMS documentation by mapping it directly to the relevant ISO 13485 clauses.16 This includes providing a dedicated QMSR module for modular PMAs and ensuring that the submission contains both procedures and “representative evidence”—such as sample process validation reports and production flow diagrams.16
The agency also now expects DUNS numbers for all manufacturing sites and a detailed plan for Unique Device Identification (UDI) system assignment and maintenance.16 Applicants must provide risk-based justifications for their cleanliness classifications, supplier controls, and validation strategies, emphasizing the rationale behind their decisions rather than just stating compliance.16
Electronic Submission and the eSTAR Platform
For electronic submissions using the eSTAR platform, the FDA has updated its requirements to ensure that the necessary QMSR information is captured in a structured format.16 This integration of the QMS review into the pre-market submission process ensures that the FDA can evaluate a manufacturer’s quality maturity long before the first pre-approval inspection (PAI) occurs.16
Economic Considerations of the Small Business Paradigm
The FDA’s economic analysis places a heavy emphasis on the impact of the QMSR on small medical device establishments. By certifying that the final rule will not have a “significant economic impact on a substantial number of small entities,” the agency is highlighting that the rule is actually a massive net positive for the small business community.34
Reduction of Compliance Effort
The “compliance effort” is defined by the agency as the time and resources required to understand, implement, and maintain a QMS that meets regulatory requirements.34 For a small firm with limited regulatory staff, the burden of maintaining both an FDA QSR-compliant system and an ISO 13485-certified system was often prohibitive.5 The QMSR allows these firms to focus their resources on a single set of requirements, which the FDA estimates will result in a net annual cost savings of over $500 million for establishments classified as small entities.34
Training and Information Technology Updates
While the long-term benefits are substantial, the FDA acknowledges that small entities will face one-time costs for training personnel and updating their documentation.34 To mitigate this, the agency provides a two-year transition period, allowing firms to spread these costs over two fiscal cycles.3 Furthermore, the agency intends to engage in variety of implementation activities, including updating information technology systems and providing training materials to help small firms navigate the new regulation.12
The Evolution of Personnel Competency and Training
The QMSR transition highlights a critical shift in how human resources are managed within a medical device quality system. Clause 6.2 of ISO 13485:2016 requires that personnel be competent on the basis of appropriate education, training, skills, and experience.14
Beyond “Reading and Signing”
In the legacy 1996 QSR, the emphasis was often on documenting that personnel had been “trained” on a specific SOP, typically evidenced by a sign-off sheet.14 The QMSR moves beyond this “reading and signing” model.14 Manufacturers must now define the required competence for each role and provide evidence that the personnel performing those roles have the necessary skills.14 This requires an assessment of training effectiveness, which could involve quizzes, practical demonstrations, or periodic performance reviews.14
Demonstrating Awareness
Additionally, ISO 13485 requires that personnel be made aware of the relevance and importance of their activities and how they contribute to the achievement of the quality objectives.32 During an inspection, an FDA investigator might interview floor-level employees to gauge their awareness of how a specific manufacturing error could impact the safety of the patient.14 This cultural awareness of quality is a key indicator of a mature QMS under the QMSR framework.2
Future Outlook: Digital Transformation and AI Regulation
As the FDA harmonizes its quality system requirements with international standards, it is also preparing for the future of medical technology, which is increasingly dominated by software, artificial intelligence (AI), and connected devices.15
SaMD and the QMSR
The QMSR is particularly well-suited for Software as a Medical Device (SaMD).13 The flexible, risk-based process approach of ISO 13485 aligns with modern software development methodologies like Agile or DevOps.33 The requirement for “Design and Development Files” allows software firms to maintain a structured history of their code iterations and algorithm updates, which is essential for managing the lifecycle of AI-driven products.13
Guidance for Artificial Intelligence-Enabled Software
The FDA’s CDRH FY2026 plan identifies guidance on artificial intelligence-enabled software as a B-list priority.36 This future guidance will likely build upon the QMSR framework, providing more specific expectations for how AI/ML models should be managed within the Medical Device File.13 Manufacturers should expect that the FDA will look for rigorous validation of AI algorithms and a robust process for managing “model drift” as part of the post-market monitoring required by Clause 8 of ISO 13485.13
The harmonization of U.S. quality systems with international standards is the first step toward a more integrated and technology-forward regulatory ecosystem.5 By adopting a common language for quality, the FDA and industry are creating the infrastructure necessary to regulate the next generation of life-saving medical innovations.4
Analysis of Global Supply Chain and Economic Operators
The adoption of ISO 13485:2016 through the QMSR also brings a heightened focus on the entire medical device supply chain.5 Clause 7.4 of ISO 13485 introduces more prescriptive requirements for the evaluation and selection of suppliers based on the risk they pose to the finished device.14
Risk-Based Supplier Evaluation
In the harmonized environment, manufacturers must move away from generic supplier approvals.27 They are expected to categorize suppliers based on their criticality (e.g., contract manufacturers, sterilizers, component suppliers) and implement appropriate levels of control and monitoring for each.27 This could include on-site audits, performance metrics, and rigorous quality agreements.27
The Role of Economic Operators
The shift also aligns the U.S. more closely with the EU MDR’s concept of “Economic Operators”.38 While the FDA’s QMSR focuses on the manufacturer, it emphasizes that outsourced processes must be controlled as part of the QMS.2 This means that distributors, importers, and service providers must be integrated into the manufacturer’s quality framework, ensuring that the safety and performance of the device are maintained from the factory door to the patient’s bedside.13
Strategic Implementation Checklist for the 2026 Transition
For organizations finalizing their transition plans, the following checklist provides a roadmap for ensuring full compliance with the QMSR by the February 2026 deadline.
- Establish a Transition Team: Appoint a cross-functional team led by senior leadership to oversee the gap analysis, documentation updates, and training programs.5
- Conduct a Formal Gap Assessment: Perform a line-by-line comparison of current procedures against ISO 13485:2016 and the specific FDA additions in the QMSR (e.g., §820.35, §820.45).5
- Develop a Quality Manual: Draft a comprehensive Quality Manual that defines the scope of the QMS and describes the interaction between its processes, as required by ISO 13485 Clause 4.2.2.26
- Build the Medical Device File Structure: Create a standard template or structure for the MDF that references all necessary specifications, manufacturing procedures, and labeling for each device family.13
- Update Risk Management Procedures: Ensure that risk management (per ISO 14971) is integrated into all facets of the QMS, including purchasing, production, and feedback loops.14
- Refine Competency Assessments: Move beyond simple training logs toward a competency-based model for all personnel performing quality-impact tasks.14
- Update Supplier Quality Agreements: Review and update quality agreements with critical suppliers to reflect the new QMSR expectations and risk-based monitoring requirements.27
- Prepare for “Management Review” Inspections: Train top management on their responsibilities under the QMSR and ensure that management review minutes are robust and inspection-ready.11
- Pilot the MDF/DDF Framework: Implement the new file structures on a new or existing product line to identify any practical challenges before the full implementation deadline.6
- Review Technical Amendments: Verify that all internal references to CFR parts (e.g., Part 801, 803, 812) have been updated to reflect the new QMSR citations.1
The Impact on Post-Market Surveillance and Adverse Event Reporting
The QMSR transition has profound implications for how manufacturers manage product performance once it enters the market. Clause 8 of ISO 13485:2016 requires the establishment of a system to provide feedback from post-market activities into the QMS.14
Proactive vs. Reactive Feedback
Traditional U.S. compliance was often reactive, focusing heavily on complaint handling and adverse event reporting (MDR).14 The QMSR, through the incorporation of ISO 13485, requires a more proactive approach.14 Manufacturers must implement methods to gather information from various sources, including customer feedback, post-market clinical follow-up (PMCF) studies, and publicly available safety data.14 This information must be systematically analyzed to identify trends and potential hazards that may not have been identified during the design phase.14
Integration with CAPA and Risk Management
The data gathered through the proactive feedback system must be integrated with the Corrective and Preventive Action (CAPA) process and the risk management process.14 If a trend in minor malfunctions is identified, the manufacturer must evaluate whether a design change or a process improvement is necessary to prevent more serious adverse events.14 This closed-loop system is essential for maintaining the “safety and performance” of the device throughout its entire lifecycle.13
Summary of Regulatory Philosophy and Future Convergence
The move to the Quality Management System Regulation is the final act in a thirty-year effort to align the United States with the global medical device community. By incorporating ISO 13485:2016 by reference, the FDA is signaling that it prioritizes harmonization, modernization, and risk-based oversight over prescriptive, region-specific regulations.4
The retirement of the DHF, DMR, and DHR in favor of the Medical Device File and the Design and Development File represents a shift toward more dynamic and integrated documentation practices.6 This shift not only reduces the administrative burden on manufacturers but also enhances the agency’s ability to ensure that devices remain safe and effective in an increasingly complex and software-driven technological landscape.12
As the February 2, 2026, compliance deadline approaches, manufacturers must embrace the challenge of QMSR implementation, recognizing that it is an investment in their future competitiveness in a global market.5 Those who successfully transition will not only satisfy the FDA but will possess a robust, internationally recognized quality management system that supports the delivery of safe and effective medical technologies to patients around the world.4
The evolution of these regulations is not a static event but an ongoing process of convergence. As the FDA continues to engage with international partners and update its guidance, the boundaries between different regulatory jurisdictions will continue to blur, ultimately leading to a more efficient, transparent, and patient-centered global regulatory ecosystem.5
Refined Conclusions and Actionable Insights for Professional Implementation
The transition from the legacy Quality System Regulation to the Quality Management System Regulation is a fundamental reconfiguration of the regulatory interface. The following conclusions and insights are derived from the totality of the analyzed regulatory shifts and are intended to guide professional implementation.
Strategic Transitioning of Documentation
The abandonment of the DMR and DHF terms necessitates a shift in documentation strategy. While the underlying data is consistent, the organization of that data must now align with the ISO 13485 “file” concept. The Medical Device File must be treated as a live repository that integrates design outputs with manufacturing specifications, providing a single source of truth for the device’s current compliance status.13
Enhancing Inspection Readiness
The removal of record exemptions for management reviews and internal audits is a major risk factor for manufacturers used to the legacy QSR.12 Organizations must implement “inspection-ready” practices for these activities, ensuring that reports are professional, findings are clearly documented, and corrective actions are tracked to completion with a rigorous assessment of effectiveness.12
Leveraging the Risk-Based Process Approach
The most significant competitive advantage of the QMSR is the mandate for risk-based thinking.2 By integrating risk management throughout the entire QMS, manufacturers can optimize their resource allocation, focusing on the most critical areas of their operations.14 This leads to more robust processes, fewer non-conformances, and ultimately, a more stable and predictable regulatory posture.12
Final Outlook on Global Harmonization
The QMSR is the bridge to a truly global MedTech market. By adopting ISO 13485, the FDA has removed one of the last major barriers to a unified global quality standard.5 Manufacturers who master the nuances of the QMSR will find themselves better equipped to launch products simultaneously in the U.S. and Europe, leveraging their Medical Device File to satisfy both FDA inspectors and EU Notified Bodies.13 This harmonization is a triumph for both industry efficiency and patient safety, ensuring that high-quality medical devices can reach the global population with fewer regulatory hurdles.4
References
- Medical Devices; Quality Management System Regulation Technical Amendments, accessed December 30, 2025, https://www.federalregister.gov/documents/2025/12/04/2025-21955/medical-devices-quality-management-system-regulation-technical-amendments
- QMSR: New FDA Regulation for Medical Devices – Efor Group, accessed December 30, 2025, https://efor-group.com/en/quality-management-system-regulation-qmsr-the-fdas-new-regulation-applicable-to-medical-devices-we-explain-it-all/
- Quality System (QS) Regulation/Medical Device Current Good Manufacturing Practices (CGMP) | FDA, accessed December 30, 2025, https://www.fda.gov/medical-devices/postmarket-requirements-devices/quality-system-qs-regulationmedical-device-current-good-manufacturing-practices-cgmp
- Medical Devices; Quality System Regulation Amendments – Federal Register, accessed December 30, 2025, https://www.federalregister.gov/documents/2024/02/02/2024-01709/medical-devices-quality-system-regulation-amendments
- Preparing for the transition from FDA QSR to QMSR – Rook Quality Systems, accessed December 30, 2025, https://rookqs.com/blog-rqs/preparing-for-the-transition-from-fda-qsr-to-qmsr
- QMSR DHF DDF Crosswalk: Key Insights and Tips – CENIT …, accessed December 30, 2025, https://cenitconsulting.com/the-no-nonsense-crosswalk-from-dhf-dmr-to-the-new-qmsr/
- DHF vs. DMR vs. DHR: Differences Explained – Greenlight Guru, accessed December 30, 2025, https://www.greenlight.guru/blog/design-history-file-dhf-device-master-record-dmr-device-history-record-dhr
- Inspection of Medical Manufacturers – 7382.845 – FDA, accessed December 30, 2025, https://www.fda.gov/media/80195/download
- FDA and ISO 13485 – Cause for Concern or Celebration? – BSI Compliance Navigator, accessed December 30, 2025, https://compliancenavigator.bsigroup.com/en/medicaldeviceblog/fda-and-iso-13485–cause-for-concern-or-celebration/
- Harmonizing ISO 13485 and FDA QSR 21 CFR 820, accessed December 30, 2025, https://13485store.com/articles/fda-qsr-shift-to-iso-13485/
- The FDA’s Proposed Quality Management System Rule: New Responsibilities and Opportunities for Medical Device Companies – Thompson, accessed December 30, 2025, https://fda.complianceexpert.com/sites/fda/files/special_report_pdf/The%20FDA%27s%20Proposed%20Quality%20Management%20System%20Rule.pdf
- Quality Management System Regulation: Final Rule Amending the Quality System Regulation – Frequently Asked Questions | FDA, accessed December 30, 2025, https://www.fda.gov/medical-devices/quality-system-qs-regulationmedical-device-current-good-manufacturing-practices-cgmp/quality-management-system-regulation-final-rule-amending-quality-system-regulation-frequently-asked
- Medical Device File (MDF) | Propel Glossary, accessed December 30, 2025, https://www.propelsoftware.com/glossary/medical-device-file-mdf
- Medtech leader Kim Trautman discusses The Medical Device Single …, accessed December 30, 2025, https://www.mddionline.com/regulatory-quality/preparing-for-2026-mdsap-as-your-regulatory-compliance-guide
- Preparing for the FDA’s QMSR: Your Guide to Compliance Success – Jama Software, accessed December 30, 2025, https://www.jamasoftware.com/blog/2025/08/19/preparing-for-the-fdas-qmsr-your-guide-to-compliance-success/
- Major Shift for QMSR Transition for Medical Device Applications …, accessed December 30, 2025, https://www.alston.com/en/insights/publications/2025/11/fda-shift-qmsr-transition-medical-devices
- DHF vs. DMR vs. DHR: Differences and Relations – SimplerQMS, accessed December 30, 2025, https://simplerqms.com/dhf-vs-dmr-vs-dhr/
- Design History File (DHF): Definition and Requirements – SimplerQMS, accessed December 30, 2025, https://simplerqms.com/design-history-file/
- What is a DHF? – Medical Device Academy, accessed December 30, 2025, https://medicaldeviceacademy.com/dhf/
- How to Create a Design History File (DHF) for Medical Devices – Ketryx, accessed December 30, 2025, https://www.ketryx.com/blog/how-to-create-a-design-history-file-dhf-for-medical-devices
- DHF Medical Device: Complete Documentation Guide – Enlil, Inc., accessed December 30, 2025, https://enlil.com/blog/dhf-medical-device-complete-documentation-guide/
- Medical Device Technical File Requirements – What you need to know – Cognidox, accessed December 30, 2025, https://www.cognidox.com/blog/medical-device-technical-file-requirements-what-you-need-to-know
- Device Master Record (DMR): Definition, Requirements, and What It Includes – SimplerQMS, accessed December 30, 2025, https://simplerqms.com/device-master-record/
- White Paper Device Master Records and Medical Device Files How Do They Compare, accessed December 30, 2025, https://www.pathwise.com/wp-content/uploads/White-Paper-Device-Master-Records-and-Medical-Device-Files-How-Do-They-Compare.pdf
- EU MDR/IVDR Technical File vs FDA Device Master Record – LFH Regulatory, accessed December 30, 2025, https://lfhregulatory.co.uk/eu-mdr-ivdr-technical-file-vs-fda-device/
- The Ultimate Comparison: ISO 13485 vs. FDA 21 CFR – OpenRegulatory, accessed December 30, 2025, https://openregulatory.com/articles/the-ultimate-comparison-iso-13485-vs-fda-21-cfr
- ISO 13485 – Medical Device Academy, accessed December 30, 2025, https://medicaldeviceacademy.com/category/13485-2016/
- Technical Documentation for MDR – WQS, accessed December 30, 2025, https://us.wqs.de/technical-documentation-mdr/
- Medical Device Technical File (Technical Documentation) – SimplerQMS, accessed December 30, 2025, https://simplerqms.com/medical-device-technical-file/
- MDR Technical Documentation Guide: Annex II & III Outline – specculo, accessed December 30, 2025, https://specculo.com/technical-files/
- Technical documentation for medical devices – Johner Institute, accessed December 30, 2025, https://blog.johner-institute.com/tag/technical-documentation-for-medical-devices/
- FDA 21 CFR Part 820 vs. ISO 13485:2016 vs. ISO 13485:2003 – Greenlight Guru, accessed December 30, 2025, https://www.greenlight.guru/blog/iso-13485-2016-iso-13485-2003-fda-21-cfr-part-820
- Readiness Checklist: Preparing for FDA’s 2026 QMSR – Veranex, accessed December 30, 2025, https://veranex.com/blog/readiness-checklist-preparing-for-fdas-2026-qmsr
- Medical Devices; Quality System Regulation Amendments – FDA, accessed December 30, 2025, https://www.fda.gov/media/176147/download
- The Clock is Ticking Are You Ready for the QMSR Challenge – NSF, accessed December 30, 2025, https://www.nsf.org/knowledge-library/ready-for-qmsr-challenge
- FDA CDRH Announces Priorities for Fiscal Year 2026, accessed December 30, 2025, https://www.emergobyul.com/news/fda-cdrh-announces-priorities-fiscal-year-2026
- FDA QMS Vs ISO 13485 – 2016-Gap Analysis-Print | PDF | Verification And Validation | Quality Management System – Scribd, accessed December 30, 2025, https://www.scribd.com/document/751119521/FDA-QMS-vs-ISO-13485-2016-Gap-analysis-print
- EU Medical Device Regulation Comparison to ISO 13485 | SGS Switzerland, accessed December 30, 2025, https://www.sgs.com/en-ch/news/2020/03/eu-medical-device-regulation-comparison-to-iso-13485